
gmx dssp
gmx dssp
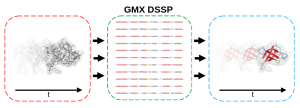
gmx dssp [1] — это инструмент для определения вторичной структуры белков в молекулярно-динамических симуляциях, основанный на алгоритме DSSP и интегрированный в пакет GROMACS. Он анализирует водородные связи между аминокислотными остатками, используя геометрические критерии, такие как электростатическая энергия или углы и расстояния между атомами. В зависимости от выбранного режима обработки атомов водорода, программа может работать с координатами водородов напрямую или использовать псевдоатомы, созданные на основе соседних атомов. Для повышения производительности расчётов применяется метод поиска соседей, рекомендующий использование радиуса отсечки не менее 0,9 нм.
Инструмент позволяет гибко настраивать распознавание различных типов вторичных структур, включая приоритетное выделение π-спиралей и параметры полипролиновых спиралей. Результаты анализа представлены в виде матриц с временным разрешением, которые можно визуализировать, а также в форме статистики по типам структур, таких как α-спирали, β-листы, изолированные мосты и изгибы. Кроме того, возможно вычисление растворимой поверхности белка, выраженной как в абсолютных значениях, так и в долях от максимума. Поддерживаются версии DSSP 1, 2 и 4, что позволяет использовать разные форматы данных.
Данный модуль объединяет точность классического алгоритма DSSP с высокой производительностью и удобством использования в GROMACS, обеспечивая детальный и надёжный анализ динамики вторичной структуры белков в биомолекулярных симуляциях.
References
- (2024): DSSP in GROMACS: Tool for Defining Secondary Structures of Proteins in Trajectories. In: Journal of Chemical Information and Modeling, vol. 64, no. 9, pp. 3593-3598, 2024.